MOLECULAR MODELING and MOLECULAR GRAPHICS.
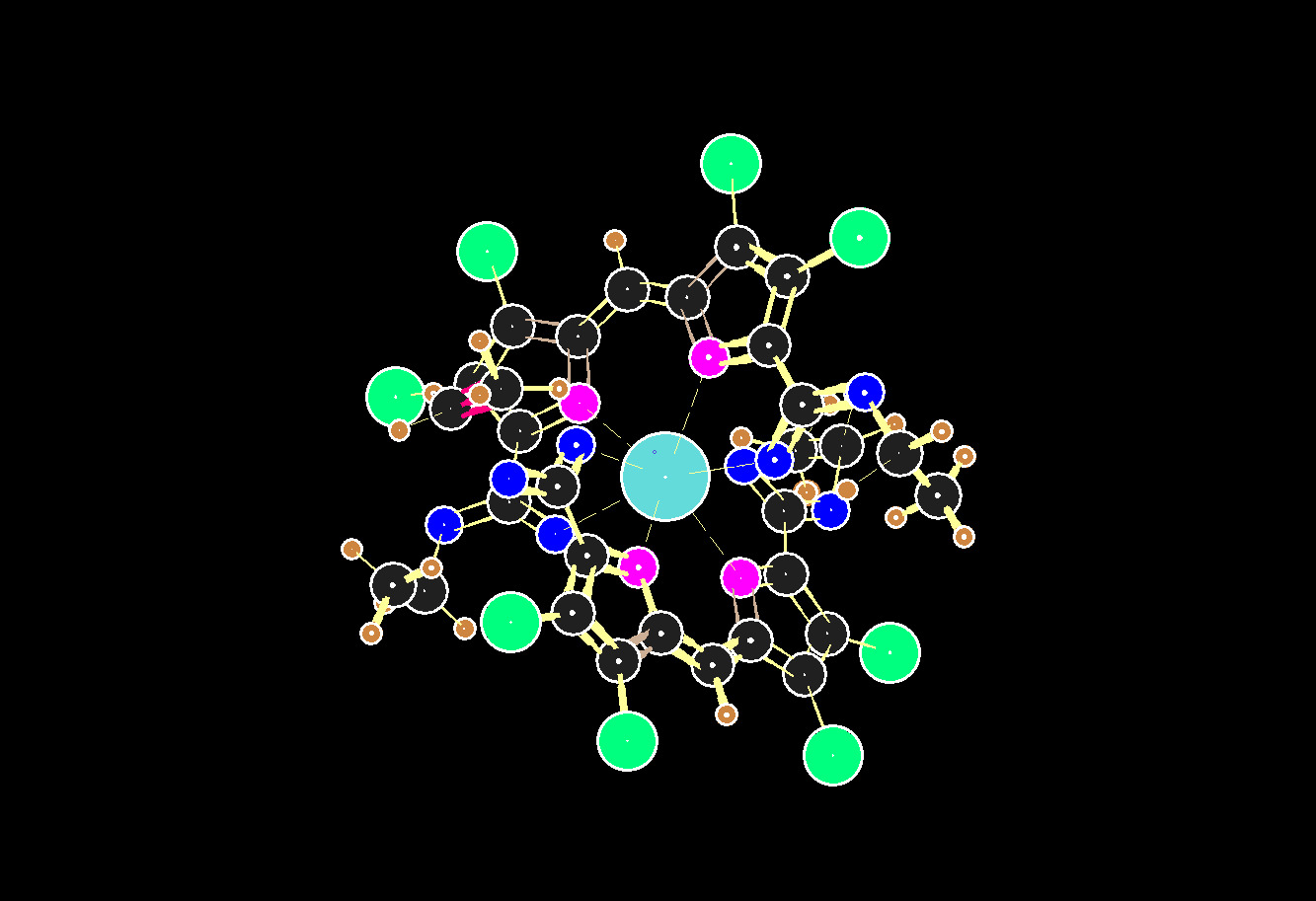
A chelated calcium ion, in the x-ray coordinate data, of JESXIO, flawed by coordinate data errors.
Find out about StruMM3D's molecular models, how you can generate them, and how you can use them in your daily tasks, presentations, reports and publications.
Can You Spot The Coordinate Data Errors?
Every now and then, whenever we find interesting X-ray coordinate date, we shall run an “awareness exercise” in which we ask readers, and users or molecular modeling software, to acquire and examine the coordinate data for selected molecules from the web-based iUCr Structure Reports, or using files that we make available and that can be downloaded, for example those below.
If the file is at the iUCR site, then simply go to the iUCr Structure Reports site, click on “search”, and put in the structure code given below. Once the hit is displayed, download the cif file, and examine the structure using your current molecular modeling software. Each structure will have one, or more, coordinate data errors.
If you do manage to spot the atomic coordinate data errors in the selected structure, write to us and let us know
1. What the errors related to, in terms of structure.
2. How long it took for you to find the error(s).
3. How difficult it was to complete the entire exercise, from downloading the data to finding the structure flaws.
We’ll publish hints as to where the flaws lie.
Examine These Flawed Structures (these are downloadable .PDB files and/or .XXS files for StruMM3D)
CLUE - Measure the lengths of the C-O bonds!